Textsuche
Needleman-Wunsch-Algorithmus
Problem
Gegeben sind zwei Proteinsequenzen, als einfaches Beispiel etwa die Sequenzen MAEHTE und AEHREN. Gefragt ist nach der Ähnlichkeit dieser Sequenzen.
Schreibt man die Sequenzen untereinander, so zeigt sich, dass sie an keiner Position übereinstimmen.
| M | A | E | H | T | E | ||
| A | E | H | R | E | N |
Tatsächlich aber sind die Sequenzen nicht grundverschieden, sondern sie enthalten gleiche Teilstücke, z.B. AEH und ein weiteres E, jedoch an verschiedener Position. Diese Übereinstimmung lässt sich verdeutlichen, indem jeweils in einer der Sequenzen Lücken (gaps) eingefügt werden.
| M | A | E | H | T | E | _ | |
| | | | | | | | | ||||
| _ | A | E | H | R | E | N |
Die Folge der an den senkrechten Strichen übereinstimmenden Buchstaben bildet die längste gemeinsame Teilsequenz (engl.: longest common subsequence) der beiden Sequenzen. Die Ausrichtung der beiden Sequenzen an der längsten gemeinsamen Teilsequenz wie oben dargestellt wird als Alignment bezeichnet.
Die Länge der längsten gemeinsamen Teilsequenz, bezogen auf die Länge der Sequenzen, ist ein Maß für die Ähnlichkeit der beiden Sequenzen. D.h. je mehr senkrechte Striche das Alignment hat, desto größer ist die Ähnlichkeit. Je mehr Nicht-Striche das Alignment dagegen hat, desto größer ist die Unterschiedlichkeit der beiden Sequenzen.
Die Anzahl der Nicht-Striche des Alignments wird als die Editier-Distanz (engl.: edit distance) der beiden Sequenzen bezeichnet. Dieses Maß gibt an, wieviele Buchstaben-Änderungen, Einfügungen und Löschungen (= Einfügungen in der anderen Sequenz) mindestens erforderlich sind, um die eine Sequenz in die andere zu überführen. Im obigen Beispiel etwa lässt sich MAEHTE in AEHREN überführen, indem das M gelöscht wird, das T in ein R geändert wird und am Schluss ein N eingefügt wird, die Editier-Distanz ist also 3.
Die Editier-Distanz ist eine Metrik, also eine Abstandsfunktion, auf der Menge der Sequenzen. Je größer der Abstand zwischen zwei Sequenzen ist, desto unterschiedlicher sind sie.
Noch genauer lässt sich das Maß der Ähnlichkeit zweier Sequenzen fassen, wenn die Übereinstimmung gewisser Buchstaben höher bewertet wird als die anderer, wenn die Nichtübereinstimmung bestimmter Buchstaben-Kombinationen anders bewertet wird als als die anderer, und wenn auch Einfügen und Löschen mit einer eigenen Bewertung versehen werden.
Im folgenden Beispiel bewerten wir zunächst der Einfachheit halber jede Übereinstimmung mit 3, jede Nichtübereinstimmung mit 0, und jede Einfügung bzw. Löschung mit -1. Hierzu werden die Variablen match, mismatch und gappenalty eingeführt.
| match | = | 3 | (engl.: match – Übereinstimmung) | |
| mismatch | = | 0 | (engl.: mismatch – Nichtübereinstimmung) | |
| gappenalty | = | -1 | (engl.: gap penalty – Strafpunkte für eine Lücke) |
Algorithmus
Mit dem folgenden Verfahren, dem Needleman-Wunsch-Algorithmus, wird zunächst das Maß der Ähnlichkeit von zwei Sequenzen und anschließend ein Alignment berechnet [NW 70].
Gegeben sind zwei Sequenzen x und y. An die Sequenzen wird jeweils zu Anfang ein spezielles Zeichen $ angefügt. Die Längen der Sequenzen danach werden mit m bzw. n bezeichnet.
Bei der Berechnung wird eine Funktion equal benutzt, die zwei Zeichen miteinander vergleicht. Der Funktionswert equal(i, j) ist gleich match, wenn das i-te Zeichen von x mit dem j-ten Zeichen von y übereinstimmt, und sonst gleich mismatch:
| equal(i, j) = | 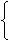 |
|
für alle i ∈ {0, ..., m-1}, j ∈ {0, ..., n-1}.
Es wird nun eine m × n-Matrix a nach folgender Vorschrift ausgefüllt:
a0, 0 = 0
a0, j = a0, j-1 + gappenalty
ai, 0 = ai-1, 0 + gappenalty
ai, j = max (ai-1, j + gappenalty, ai, j-1 + gappenalty, ai-1, j-1 + equal(i, j) )
für alle i ∈ {1, ..., m-1}, j ∈ {1, ..., n-1}.
Bei der Berechnung der Einträge der Matrix wird also auf vorher berechnete andere Einträge zurückgegriffen; diese Methode der Berechnung bezeichnet man als dynamische Programmierung (engl.: dynamic programming) .
.
Für das Beispiel ergibt sich folgende Matrix:
| $ | A | E | H | R | E | N | |
|---|---|---|---|---|---|---|---|
| $ | 0 | -1 | -2 | -3 | -4 | -5 | -6 |
| M | -1 | 0 | -1 | -2 | -3 | -4 | -5 |
| A | -2 | 2 | 1 | 0 | -1 | -2 | -3 |
| E | -3 | 1 | 5 | 4 | 3 | 2 | 1 |
| H | -4 | 0 | 4 | 8 | 7 | 6 | 5 |
| T | -5 | -1 | 3 | 7 | 8 | 7 | 6 |
| E | -6 | -2 | 2 | 6 | 7 | 11 | 10 |
Die Zahl in der rechten unteren Ecke (im Beispiel die 10) ist das Maß für die Ähnlichkeit der beiden Sequenzen.
Ein Alignment der beiden Sequenzen lässt sich konstruieren, indem ausgehend vom rechten unteren Eckfeld immer in der Richtung gegangen wird, aus der das Maximum gekommen ist. Die entsprechenden Felder sind in der obigen Matrix fett gekennzeichnet. Wird dabei nach links gegangen, entspricht dies einer Einfügung in der Sequenz x, wird nach oben gegangen, entspricht dies einer Einfügung in der Sequenz y. Wird diagonal nach links oben gegangen, entspricht dies einer Übereinstimmung, wenn der Wert des aktuellen Feldes und der Wert des linken oberen Nachbarfeldes sich um match unterscheiden, und sonst einer Nichtübereinstimmung. So kommt das folgende Alignment zustande.
| x: | M | A | E | H | T | E | _ | ||
| | | | | | | | | ||||||
| y: | _ | A | E | H | R | E | N |
Implementierung
Die zu berechnende Matrix belegt einen Speicherplatz von Θ(m·n). Wenn m und n groß werden, ist dies problematisch. Zur Berechnung der Editier-Distanz, also der Zahl in der unteren Ecke der Matrix, genügt offenbar auch Θ(n) Speicherplatz, wenn die Matrix zeilenweise berechnet wird und immer nur die jeweils zuletzt berechnete Zeile gespeichert wird. Jedoch lässt sich das Alignment dann nicht mehr in der angegebenen Weise konstruieren.
Es gibt jedoch den Algorithmus von Hirschberg, der dieses Problem durch eine modifizierte Art der Berechnung löst [Hir 75].
Literatur
[HD 06] M.T. Hütt, M. Dehnert: Methoden der Bioinformatik. Springer (2006)
[NW 70] S.B. Needleman, C.D. Wunsch: A general method applicable to the search for similarities in the amino acid sequence of two proteins. Journal of Molecular Biology 48, 3, 443-453 (1970)
[Hir 75] D.S. Hirschberg: A linear space algorithm for computing maximal common subsequences. Communications of the ACM 18, 6, 341-343 (1975)
Weiter mit: [up]
H.W. Lang mail@hwlang.de Impressum Datenschutz
Created: 15.09.2006 Updated: 13.03.2023
Diese Webseiten sind während meiner Lehrtätigkeit an der Hochschule Flensburg entstanden